
|
Source: SEER Data (2018)
A musical composition based on algorithms derived from breathing patterns and ECG/EEG waveforms2/22/2018
For my medical school thesis in 2002, I wrote a composition that was based on algorithms derived from waveforms reflecting hypoventilatory breathing, various cardiac rhythms on ECGs, and sleep patterns captured on EEGs. The hypothesis was that every human condition, from the balanced healthy state in homeostasis to terminal derangement manifested by events such as agonal breathing and cardiac arrhythmias, occurs in measurable mathematical units that can be used to compose a multi-instrumental composition in harmonious segments and specific keys corresponding to each physiological state. Under this hypothesis, conditions representing different physiological states (e.g., disease vs healthy states) may be in different keys but should be mathematically related. Each state would have a center of gravity (key) and the transition from the healthy state to disease can be heard as a change in key or time signature.
The practical implication of this thesis is as follows: once a human state is quantified, any deviation indicative of signs and symptoms of disease would appear in a musical composition as a dissonant tone and out of sync with the baseline. Therefore, machine learning and artificial intelligence algorithms trained to learn these mathematical ratios and metronomic patterns can potentially predict the probability and timing of transitioning from one state to another (e.g. from the healthy state to disease and from a disease state to death). In some cases, these transition states could be detectable by the machine, or easily heard in a musical composition as dissonant anomalies, before signs and symptoms of disease are apparent to a diagnostician. Dissecting out the composition, perpetual drift The composition starts with agonal breathing and hypoventilatory gasp superimposed on a beating heart (S1 and S2 beats) in the setting of a second degree heart block. 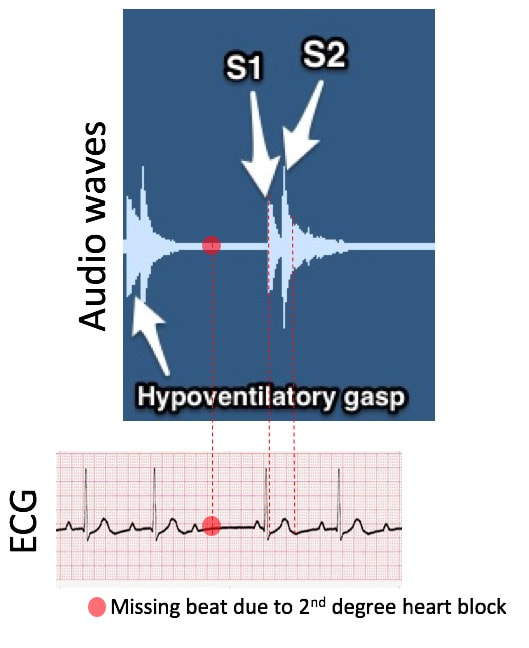
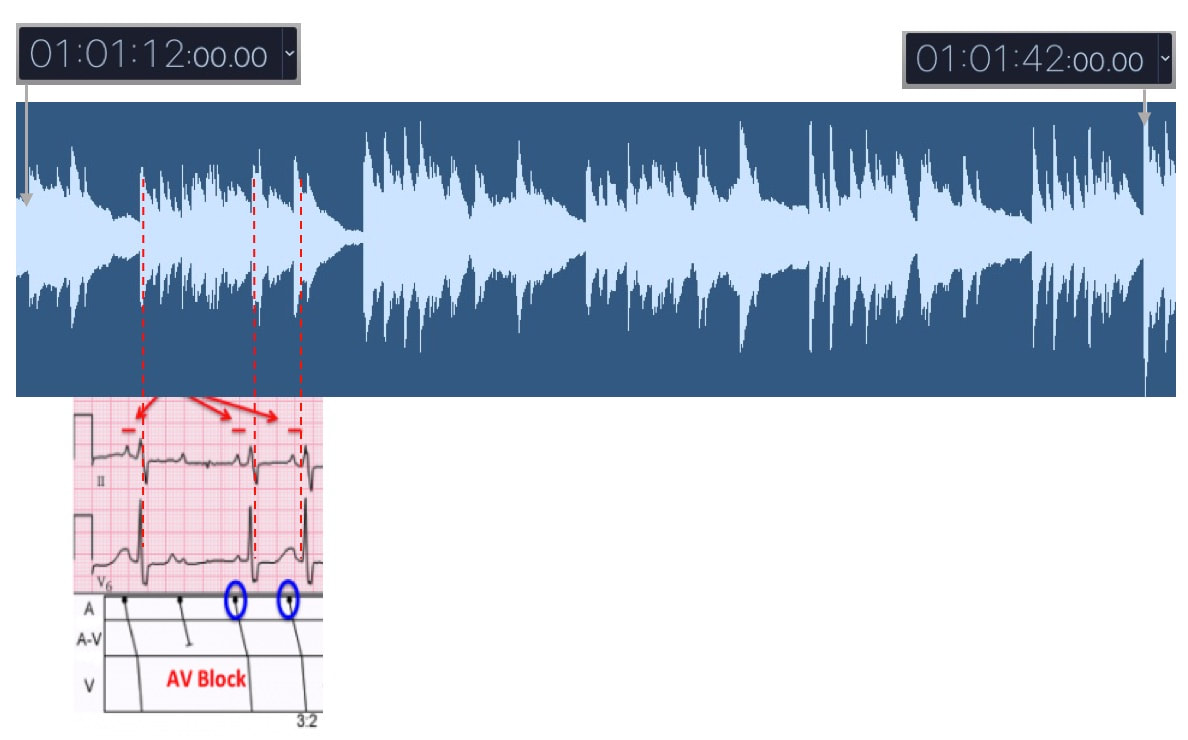
From time point 1:01:12 to 1:01:42, the ECG rhythm degrades to a higher degree atrioventricular (AV) block:
Bursts of polymorphic ventricular tachycardia (PVT) are heard about 2:33 minutes into the composition. The QRS complexes during PVT are characterized by varying amplitude, axis, and duration; creating musical dissonance as the physiological state transitions into a near terminal state. This dissonance is clearly heard when the vocalist continues to sing in the original key and time signature around minute 3:30 and 3:53.
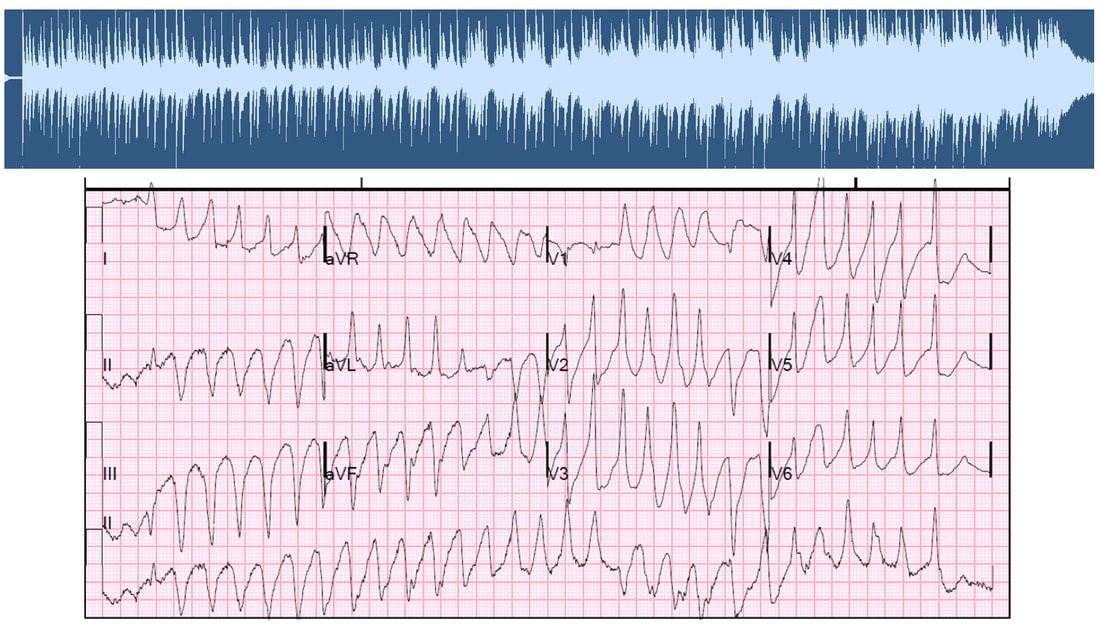
Audio waves (top) based on polymorphic ventricular tachycardia waveforms on ECG (bottom)
The composition ends on a happy note with a classical guitar playing chords based on EEG alpha waves that typically occur during a relaxed wakeful state. The story in perpetual drift could've all been a dream.
Recorded in 2002. Originally released by Azure. Composer: Sean Khozin Vocals: S. Gunn Special thanks to my med school mentors who tolerated and supported this project.
I’d like to thank Dr. Abrahams and members of the organizing committee for inviting me to speak here today. This conference is a great testament to the progress of recent years that has catalyzed major leaps in advancing the public health. We’re all deeply aware that we’re nearing an inflection point in the life sciences, with unprecedented opportunities for developing a new generation of highly personalized therapies.
Personalized medicine started to appear in the English literature in the late 1800s, and has been a driving force behind major advances in biomedicine ever since. In the early days, personalization of care was largely the function of the therapeutic doctor-patient relationship. For example, up until the mid 20th century, country doc’s strategy of making house calls was a common method for delivering medical care that incorporated quantitative information, such as vital signs, and qualitative evidence such as a patient’s performance and socioeconomic status into developing individualized care plans. Modern concepts in personalized medicine can best be defined by their focus on utilizing advances in technology to optimize personalization and delivery of care. 
Country Doctor, by W. Eugene Smith
Blood typing to guide transfusions and predicting hypersensitivity reactions to antiretroviral regimens based on the presence of specific alleles were preludes to a biomarker driven approach to personalization of care that today are bringing us precision therapies targeted to genomic drivers of disease initiation and maintenance. FDA has been highly responsive and adaptive to this rapidly changing landscape of drug development, leveraging its regulatory authority to enable diffusion of innovations into the market while maintaining appropriate standards for product quality, integrity, safety, and effectiveness to protect patients and consumers of healthcare goods and services.
FDA’s expedited programs have created an efficient regulatory framework for accelerating the development of personalized drugs and biologics for serious conditions that pose the greatest burden on patients and society. Fast track designation and accelerated approval have supported numerous regulatory actions to streamline clinical development programs, providing timely access to potentially lifesaving therapies. Breakthrough therapy designation, the most recent addition to FDA’s menu of expedited programs, has provided a uniquely collaborative regulatory context for speeding up the development of personalized therapies based on early evidence of substantial clinical activity. Following breakthrough therapy designation, FDA mobilizes its internal resources to deliver timely advice to help sponsors design and conduct a drug development program as efficiently as possible. Co-development of companion diagnostic assays are tightly coordinated between sponsors and FDA’s devise and drugs or biologics divisions for concurrent approval decisions. In helping innovators achieve clinical development goals for breakthrough therapy designated products, FDA is open to alternative trial designs such as adaptive, N of 1s, and real world data based studies. The success of the breakthrough therapy designation program has been clearly demonstrated through regulatory actions such as the recent approval of a small molecule for the treatment of patients with EGFR T790M-positive non-small cell lung cancer. This drug was approved only 2.5 years following the sponsor’s submission of the initial investigational new drug application for the first in human phase 1 clinical trial. The days of decade long clinical development programs are indeed numbered, thanks to the kind of work that many of you sitting here have pioneered and advocated. The technological and scientific advances that are paving the way for a new generation of personalized therapies are inspiring us to develop new methods of generating evidence and delivering care. Our clinical evidence generation system, largely based on the gold standard of controlled empiricism pioneered by James Lind in the famous scurvy trial of 1747 has served us very well. The processes and procedures governing the conduct of conventional clinical trials derived from the principles of controlled empiricism have ensured internally valid study designs, reducing bias and controlling for alternative explanations of treatment effects. Since James Lind’s bold experiment in 1747, we’ve made great progress in improving the ethical standards of clinical research, primarily in response to events during World War II. However, progress in developing innovative ways of conducting clinical trials has been lagging, making the existing methods somewhat outdated and inefficient. Conventional clinical trials capture the experience of a carefully curated population of patients with results that sometimes do not support personalized treatment decisions at the point of care. The results of many conventional clinical trials, typically reported in average treatment effects, are often geared towards broad generalization, as opposed to personalization. Over-controlled experimental conditions such as narrow patient enrollment criteria, have reduced the external validity of conventional trials, hindering our ability to make accurate assumptions about personalized treatment decisions. Evidence for high value interventions and information on large segments of the population, such as patients with commodities, the elderly, and minorities is sometimes lacking. It should, therefore, come as no surprise that clinicians still rely on clinical judgement, opinion, and anecdotal evidence for a significant number of treatment decisions today, despite diligent efforts to develop a more personalized therapeutic armamentarium. By leveraging the rapidly evolving opportunities enabled by data science and technology, including digital health solutions and real world evidence, we can address the external validity deficit of conventional clinical trials and improve clinical evidence generation, making it more patient-centered and personalized. 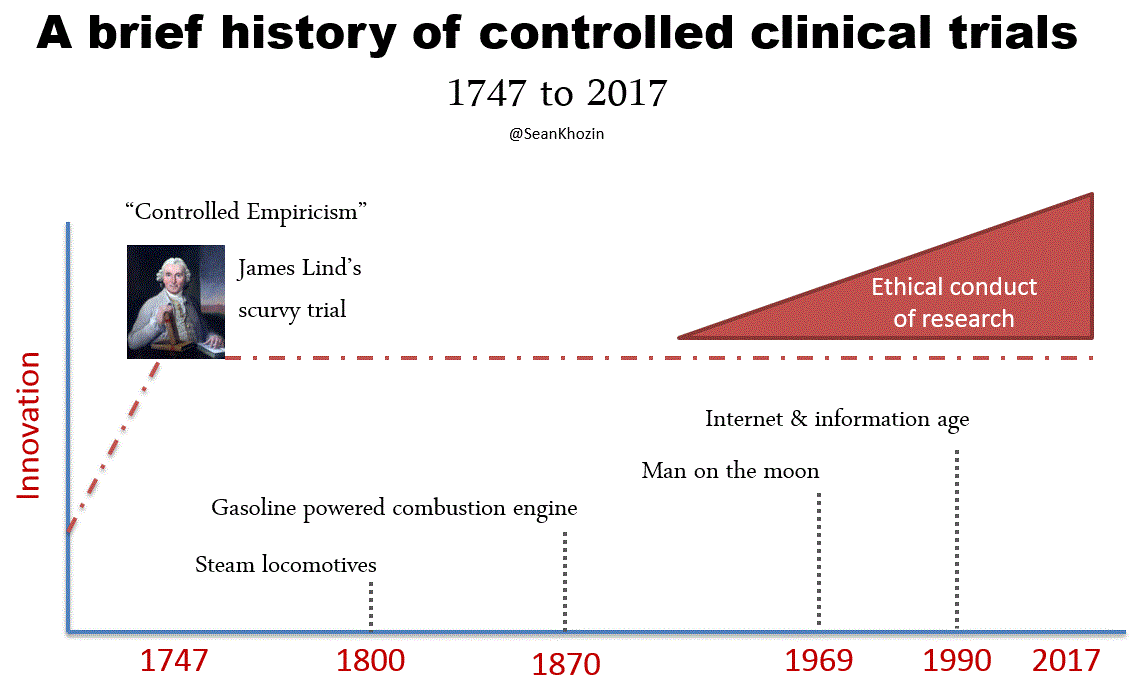
Little innovation in clinical evidence generation since Jame Lind introduced the principle of "controlled empiricism" following his landmark scurvey trial in 1747, despite great technological advances: from steam locomotives to the information age. Significant progress in ethical conduct of research was made following events of WWII.
In addressing these challenges and opportunities, FDA is developing new regulatory frameworks and has started several pilots and demonstration projects focused on biomedical innovation and modernizing clinical evidence generation. For example, an oncology data science and health technology incubator called Information Exchange and Data Transformation (INFORMED) at FDA Oncology Center of Excellence is building organizational and technical capacity for big data analytics and development of modern approaches to evidence generation to help inform regulatory decisions. As a collaborative sandbox with a large portfolio of research collaborations and public-private partnerships, INFORMED has launched three demonstration projects related to the use of real world data for evidence generation. Two of these projects are investigating the use of data from electronic health records collected as part of routine delivery of care, to examine the experience of cancer patients not typically represented in conventional clinical trials. A third project is a clinical study piloted in collaboration with the National Cancer Institute aimed at developing new methods based on biometric sensors for estimating the functional and cognitive status of patients with advanced malignancies undergoing treatment. Other efforts in the portfolio include projects testing the utility of artificial intelligence and machine vision algorithms to help streamline drug development programs.
We all recognize that personalization of medicine is a data intensive effort, at the heart of which lies collection and analysis of a wide variety of data types including genomic, proteomic, and microbiome based pipelines, to characterize intrinsic and extrinsic variables influencing the patient’s experience and response to therapies. FDA is facilitating responsible sharing of health data to create new mechanisms for generation of insights using predictive analytics, open science, and crowdsourced solutions. Among these are efforts in collaboration with academia, nonprofits and patient advocacy groups to develop external control arms by pooling data from relevant clinical trials and electronic health records. External control arms from properly curated and validated data lakes could serve as a reliable comparative benchmark for a product showing early evidence of clinical activity in a single arm trial, potentially obviating the need to conduct conventional randomized studies, especially in situations where equipoise is compromised. FDA is also prototyping a data sharing framework based on blockchain that may facilitate a decentralized and secure method of sharing data among institutions and most importantly, by individual patients, who currently lack efficient means of sharing their health data in a reliable, secure, and scalable manner. 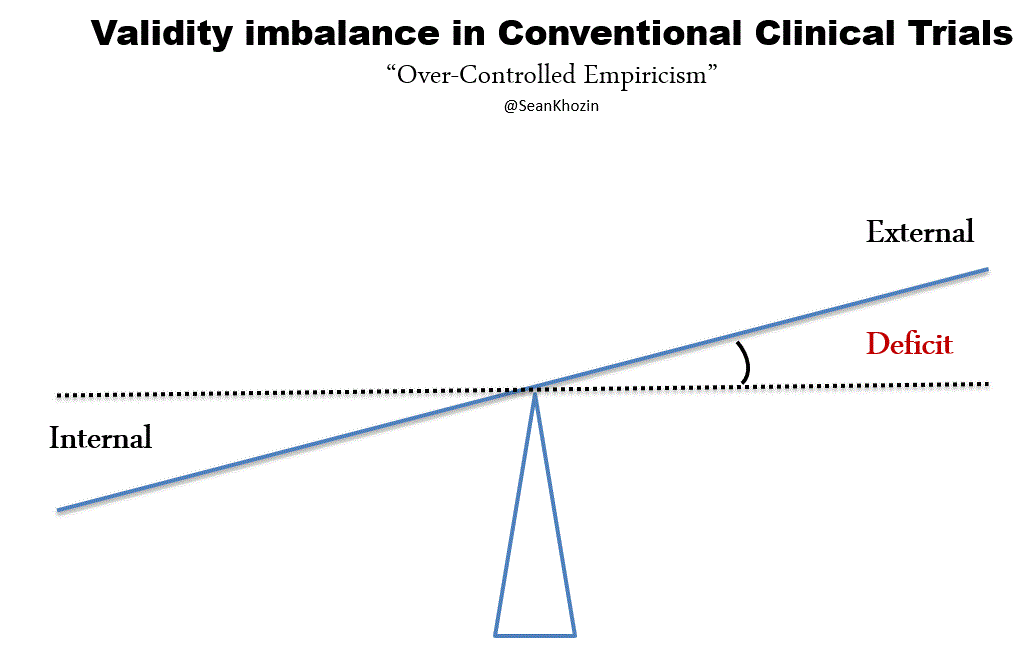
Appropriately-designed real world studies can address external validity defecits in conventional clinical trials
We’re in a brave new world presenting us with great new opportunities to improve the public health. Long gone are the days of reliance on tactile palpation and auscultation as the primary means of data collection in clinical trials. Increasingly, machines and devices are collecting and generating large volumes and near continuous streams of data directing clinical development decisions. Ensuring the analytical validity of these devices has been at the forefront of FDA’s efforts in recent years, from the days of companion diagnostic assays to the most recent efforts articulated in the agency’s new Digital Health Innovation Action Plan. The genesis of this plan was based on the recognition that traditional methods of regulating medical devices may not be suitable for the development and validation of modern digital health solutions that are key components of personalization of care in the 21st century. For example, in a recent randomized clinical trial involving 766 patients with metastatic solid tumors, a simple web-based questionnaire designed to track 12 common side effects related to anticancer therapy was associated with a 5-month improvement in overall survival; an efficacy margin of unquestionable breakthrough status. FDA’s Digital Health Innovation Action Plan contains an important framework called the Pre-Certification Program representing a modern approach to regulation, one that shifts the focus from the product to the developer of the product. We’re examining the application of the Pre-Certification program, where the FDA becomes a certifier of device manufacturers seeking to go to market, to other areas in our regulatory portfolio. For example, this construct can potentially inform the development of a modern legislative approach to regulation of laboratory-developed tests.
FDA’s mission of promoting and protecting the public health is centered on the assurance of efficiency, transparency, and consistency in the regulatory process, so that safe and effective therapies can reach patients that need them. As we enter what some call the 4th industrial revolution, we can all benefit from re-evaluating our workflows and conventional means of doing business through systems reengineering powered by smart technologies and a collaborative approach to drug development that turns a keen eye to where therapies and medical interventions are personalized: the point of routine care. Real world data, digital health, next gen diagnostics, artificial intelligence and automation are all part of the new realities and opportunities in drug development. These innovations can greatly streamline the mechanics of the enterprise, where significant resources are currently being devoted, so that the sampling frame is recalibrated to addressing the needs and understanding the experiences of individual patients: like a modern-day country doc obsessed with making house calls to personalize the care experience. Thank you for the opportunity to be here today. |




 RSS Feed
RSS Feed